
一
AI医疗影像技术的发展革新
随着人工智能(Artificial Intelligence,简称“AI”)技术的迅速发展尤其对各种应用场景的适配性提高,AI技术正以前所未有的速度改变着医疗行业,其中即包括医疗影像。AI医学影像主要用于医疗健康市场和大健康场景中,在医疗健康市场中主要用于协助医生进行疾病监测及诊断,大健康场景中主要用于健康风险评估。AI赋能于医疗影像具有多方面优势,首先,AI可先行初步处理处理和分析医学影像数据,加速人工诊疗程序,显著提高了人工的诊断工作效率;其次,通过AI模型训练后,可以客观识别并捕捉影像中的细微变化和特点,减少人工主观判断的偏差而提高诊断的准确性。
二
AI医疗影像技术的现状及法律挑战
相比较于Deepseek近期的商业化,AI赋能在医疗影像领域的适用已属于相对成熟的领域。截至目前,国内已多家企业投身于AI医学影像行业,根据市场公开信息,中国已有数十款人工智能医学影像辅助诊断软件获批国家药品监督管理局(National Medical Products Administration,简称“NMPA”)三类医疗器械证。
然而,AI医疗影像技术产品在研发、生产、经营过程中也面临多种合规及法律风险,本文拟对AI医疗影像技术软件的医疗器械注册问题进行分析和梳理,希望能为读者提供些许参考和帮助。
三
AI医疗影像系统在医疗器械法律法规下的合规要求
(一)AI影像系统设备
在实践中,AI医疗影像系统的生产及经营往往采取设备(硬件)及系统(软件)相结合的方式。类似于一般性医疗器械设备,AI医疗影像系统的设备应符合《医疗器械监督管理条例》等法律法规的监管要求。
根据《医疗器械监督管理条例》的规定,医疗器械指直接或者间接用于人体的仪器、设备、器具、体外诊断试剂及校准物、材料以及其他类似或者相关的物品,并按照风险程度高低实行三类分类管理。参考2024年版《医疗器械分类目录》,人工智能相关产品均列为第二类或第三类医疗器械进行监管。
(二)AI医疗影像系统软件
如上文所述,AI医疗影像系统为设备及软件的结合,《医疗器械监督管理条例》也将医疗器械所需要的计算机软件纳入医疗器械的监管范畴。除前述外,针对AI医疗影像系统领域,我们梳理了目前我国AI医疗器械领域的相关法规及政策汇总,请详见附件(点击文末“阅读原文”即可查看)。
1. AI医疗影像系统软件是否需注册为医疗器械
AI医疗影像系统软件是否属于医疗器械管理主要取决于软件处理对象、核心功能以及是否存在医疗用途。
根据国家药监局于2021年7月1日公布并施行《人工智能医用软件产品分类界定指导原则》,若软件产品的处理对象为医疗器械数据,且核心功能是对医疗器械数据的处理、测量、模型计算、分析等,并用于医疗用途的,符合《医疗器械监督管理条例》有关医疗器械定义,作为医疗器械管理。若软件产品的处理对象为非医疗器械数据(如患者主诉等信息、检验检查报告结论),或者其核心功能不是对医疗器械数据进行处理、测量、模型计算、分析,或者不用于医疗用途的,不作为医疗器械管理。
经查询公示信息,市场上陆续在心血管疾病、肺结节、乳腺疾病等领域推出AI医学影像软件并获批的三类医疗器械,如“颅内动脉瘤手术计划软件”“冠状动脉CT血流储备分数计算软件”“胸部CT图像处理与分析软件”“乳腺X射线图像辅助检测软件”等,均已注册为医疗器械。
2. AI医疗影像系统软件是否需要单独注册
AI医疗影像系统软件是否需要单独注册取决于是否为独立软件(SaMD)。
根据《医疗器械软件注册审查指导原则》,独立软件(SaMD)和软件组件(SiMD)的区分如下图所示:
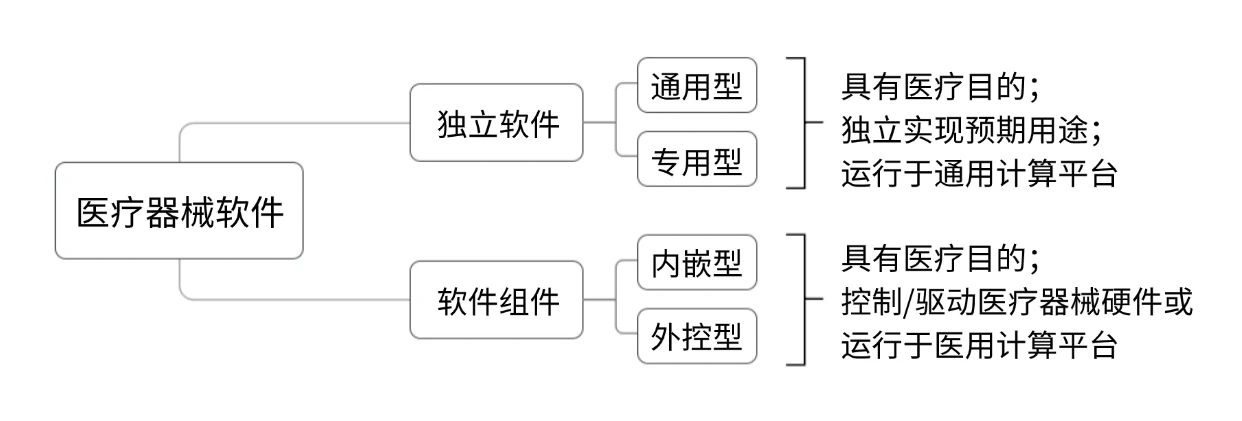
常见的独立软件包括医学图像处理软件、患者监护软件、动态心电数据分析软件等;常见的软件组件包括脑电图机所含嵌入式软件(即固件)、CT、MRI图像采集工作站软件等。
▶对于独立软件:以单独注册为原则,作为附件注册为例外。根据《医疗器械软件注册审查指导原则》要求,独立软件作为医疗器械或医疗器械附件,通常单独注册,特殊情况可随医疗器械进行注册,此时虽不控制/驱动医疗器械硬件但从产品角度运行于医用计算平台,故视为软件组件,如专用型独立软件可作为附件随医疗器械进行注册。
▶对于软件组件:一般而言无需单独注册。软件组件作为医疗器械或医疗器械部件、附件的组成部分,不宜单独注册,需随医疗器械进行整体注册。
经查询公开案例,Airdoc-AIFUNDUS(1.0)作为一款用于辅助诊断糖尿病视网膜病变的人工智能医疗器械软件(SaMD),已获国家药监局第三类医疗器械注册。
3. AI医疗影像系统软件的管理分类
如AI医疗影像系统软件需进行单独注册,需判断其适用二类或三类的管理标准,划分标准主要取决于医疗影像系统软件的适用范围。
根据《医疗器械分类目录》《人工智能医用软件产品分类界定指导原则》等要求及器审中心的答疑回复,对于算法在医疗应用中成熟度低(指未上市或安全有效性尚未得到充分证实)的人工智能医用软件,若用于辅助决策,如提供病灶特征识别、病变性质判定、用药指导、治疗计划制定等临床诊疗建议,按照第三类医疗器械管理;若用于非辅助决策,如进行数据处理和测量等提供临床参考信息,按照第二类医疗器械管理,若还是不能明确管理类别的,则建议注册申请人申请分类界定。
经查询NMPA公示信息,对于注册为二类的医疗器械软件产品,其适用范围多包括数据导入、显示、浏览和数据计算等;对于注册为三类的医疗器械软件产品,其适用范围多为包括病灶识别、数据分析等。值得注意的是,部分注册为三类的医疗器械在适用范围中明确注明了供经培训合格的医师临床使用,不能单独作为临床诊疗决策的依据。
(三)AI医疗影像系统的境外合规
医疗器械进入国际市场时,需符合进口国医疗器械相关法律法规要求。国际上主要的医疗器械市场所在的国家和地区普遍按照分类监管的原则对医疗器械实施管理。
欧美日等发达国家地区依旧为我国医疗器械主要出口市场,据中国医药保健品进出口商会数据统计,2024年我国医疗器械的出口市场前三位分别为美国(出口额117.6亿美元,占比24.1%)、日本(出口额27亿美美元,占比5.5%)、德国(出口额25.9亿美元,占比5.3%)。
▶美国FDA监管
美国医疗器械监管机构为食品药品监督管理局(Food and Drug Administration,简称“FDA”)。
根据the Federal Food, Drug, and Cosmetic Act(简称“FD&C法案”)第201(h)(1)条规定,医疗器械指:仪器、仪表、器具、设备、装置、植入物、体外诊断产品或其他类似或相关的物品,包括以下产品或附件:(1)收录在国家处方集、美国药典或其补充性文件;(2)拟用于诊断疾病或其他病症,或用于治愈、减轻、治疗或预防人类或其他动物的疾病;或(3)旨在影响人体或其他动物身体的结构或任何功能,并且不能通过人体或其他动物体内或体外的化学作用达到其主要预期目的,并且不依赖于代谢来实现其主要预期目的。术语“设备”不包括根据第520(o)条排除的软件功能。
美国对医疗器械实行分类管理,根据其风险程度分为三类(I类、II类或III类),其中III类器械受到最严格的监管控制。根据我们查询公开信息,在美国AI医疗影像系统软件多为II类或III类,适用510(k)、De Nove或PMA的监管方式。
▶日本MHLW监管
日本医疗器械的监管机构为厚生劳动省(Ministry of Health, Labor and Welfare,简称“MHLW”)。根据《药品与医疗器械法》(Pharmaceutical and Medical Device Act,简称“PMD Act”)第2条第4项,医疗器械定义为:用于诊断、治疗或预防人类或动物疾病或影响人类或动物身体结构或功能的试剂、耗材和设备。
日本对医疗器械实行分类管理,根据其对人类造成的危险程度分为一般医疗器械(Class I)、管理医疗器械(Class II)和高度管理医疗器械(Class III, IV)。根据我们查询公开信息,在日本AI医疗器械通常被列为第二类、第三类或第四类医疗器械。
▶欧盟MDR监管
欧盟主管当局及公告机构共同负责欧盟医疗器械的监督管理。根据医疗器械法规(MDR, REGULATION (EU) 2017/745),医疗器械包括用于人类仪器、器具、设备、软件、植入物、试剂材料其他物品,其预期使用由制造商确定,不论单独使用或组合使用,以达到疾病、损伤或残疾的诊断、检测等目的[1]。
欧盟对医疗器械实行分类管理,根据产品的风险等级分为I类(普通I类及Is、lm、Irs),IIa类,IIb类,III类。根据我们查询公开信息,在欧盟AI医疗器械主要被列为IIa类或更高风险分类。
另外需提请注意的是,除上述各国或地区所列监管措施外,随着AI技术的迅速推广和落地,各国各地区针对AI赋能的医疗器械(包括软件)均密集出台了相关法案、计划等具体规定,为确保合规性,建议在具体申请备案过程中随时关注监管措施的最新动态。
四
总结
随着AI技术的迅猛发展,AI医疗影像技术再次成为各界关注的焦点。近年来,我国AI医学影像市场规模持续扩大,众多企业纷纷布局,多款产品已获NMPA二类或三类医疗器械证。在监管层面,AI影像系统需遵循严格的医疗器械注册规定。如考虑到全球性布局AI医疗器械的出口亦需关注主要出口国家如美国FDA、欧盟MDR及日本MHLW等要求。面对AI技术的全面机遇与挑战,行业及各方参与者应密切关注注册要求及政策法规的动态更新,以保障合规运营。
注释

