
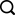
一、药品上市许可转让整体安排及考量因素
2019年12月1日,《中华人民共和国药品管理法》(2019修订)施行并确立了我国药品上市许可持有人(以下简称“MAH”)制度,我国大部分药品的上市许可和生产许可自此正式解绑,这一制度的灵活性极大地激发了药品权益交易市场活力。据统计,截至2023年10月30日,全国纯药品生产许可证B证企业(即仅委托第三方生产药品)已超过1,100家。[1]与此同时,配套MAH制度实施的《药品注册管理办法(2020)》《药品生产监督管理办法(2020)》《药品经营和使用质量监督管理办法》《药品上市后变更管理办法(试行)》《国家药监局关于加强药品上市许可持有人委托生产监督管理工作的公告》(国家药品监督管理局公告2023年第132号)等规章制度,也提出了落实药品全生命周期管理、落实药品质量主体责任的更多监管要求。
考虑到上述监管要求,在筹划药品上市许可转让安排时,需要将药品上市许可转让、生产、供应、销售和商业化等全生命周期的各环节的成本收益、法律风险等因素纳入考虑,择优选取合作模式。有鉴于此,我们结合过往多个药品上市许可转让项目经验,将各环节的主要合作模式及考量因素梳理如下:
需特别说明的是,如无特别说明,在下文及下表中,
(1)药品上市许可转让中主要涉及的主体以转让方、受让方、受托生产企业指代,转让标的以药品、产品或药品权益、产品权益指代;
(2)可能需签署的交易文件以MAH权益转让协议、技术研发委托协议、委托生产协议、委托生产质量协议(以下简称“一揽子协议”)指代;
(3)由于药品上市许可转让的转让标的一般已至少进入药品申请注册审评阶段,在合作环节中不再就临床前研究、临床试验等研发安排进行探讨。


二、药品上市许可转让基本审批/变更流程
受限于药品上市许可转让各环节的合作模式、药品特点等因素,其对应的转让基本审批/变更流程存在一定差异。就非受到特殊管理且已取得药品注册证书的药品而言,转让方和受让方签署MAH权益转让协议及其他一揽子协议后需履行的转让基本审批/变更程序如下:
1.受让方向省级药品监督管理部门申请取得或变更药品生产许可证(以下简称“取得生产许可证”)
(1)受让方应根据药品生产环节的合作模式确定应当取得生产许可证的类别,如受让方自行生产药品,则应当取得A证;如受让方委托转让方或其他第三方生产药品,则应当取得B证。[12]
(2)为取得生产许可证,受让方应当具备相应的人员(企业负责人、生产负责人、质量负责人、质量受权人等)、保证药品质量的规章制度(质量管理体系文件、药品生产风险管理制度及程序、药物警戒体系文件等),并符合药品生产质量管理规范(以下简称“GMP”)的其他要求。
(3)受让方具备申报相应类别的生产许可证条件后,可对照所在地政务服务网就办理药品生产许可证核发或变更事宜公布的办事指南,准备相应的申请资料,并向省级药品监督管理部门申请取得生产许可证。
(4)药品生产许可证核发的法定办结时限为30个工作日,许可事项变更的法定办结时限为15个工作日。
2.受让方向国家药品监督管理局药品审评中心(以下简称“CDE”)申请变更上市许可持有人
(1)申请变更上市许可持有人的程序主要分为内转内、外转外及外转内三类。[13]其中,内转内、外转外为上市许可持有人的变更;外转内为已在境内上市的境外生产药品转移至境内生产,需重新进行仿制药上市的审批。因此,外转内为严格意义上的仿制药注册上市,以下仅就内转内、外转外的流程进行阐述:
受让方向CDE提出补充申请,补充申请资料主要包括:
►药品注册证书等复印件,包括申报药品历次获得的批准文件(药品注册证书、药品补充申请批件、药品再注册批件),相应文件应当能够清晰说明该品种完整的历史演变过程和目前状况;
►证明性文件,境内生产药品,应当提交有关变更前后药品上市许可持有人的《药品生产许可证》及其变更记录页、营业执照的复印件,以及药品上市许可持有人变更协议原件(涉及商业秘密的应当隐去);境外生产药品,境外持有人指定中国境内的企业代理相关药品注册事项的,应当提供授权委托文书及公证、认证文书,并附中文译本;中国境内注册代理机构的营业执照复印件;还应当提交有关国家或地区主管部门出具的允许药品上市许可持有人变更的证明文件,以及公证、认证文书,并附中文译本。
►受让方还应就拟转让药品的生产场地、处方、生产工艺、质量标准等应当与原药品一致、不发生变更作出承诺。
CDE将会在20个工作日内完成上市许可持有人变更的审批,同意变更的,核发药品补充申请通知书,药品批准文号和证书有效期不变。
(2)如药品在申请注册审评期间发生药品注册申请人主体变更的,受让方应当向CDE提出补充申请,该申请与药品上市许可申请关联,一并送局审批,原上市许可申请审评时限不变。相关变更申请资料参照(1)中所述。[14]
3.受让方向CDE或省级药品监督管理部门申请变更生产场地、处方、工艺、质量标准、包装等(如需)
(1)如根据药品上市许可转让各环节的合作模式,需要变更原已批准的生产场地、处方、工艺、质量标准、包装等的,受让方应在完成上市许可持有人变更后,进行充分研究、评估和必要的验证,并按规定向CDE或省级药品监督管理部门申请批准、备案后实施或报告。
(2)如受让方确实需要合并申报上市许可持有人变更和生产场地等变更的,也可以合并申报,但审评时限将延长。
4.GMP符合性检查及出厂、上市放行
由于上市许可持有人变更将致使药品质量管理体系发生变更,因此,转让后的药品需要通过GMP符合性检查,满足生产企业的出厂放行及受让方的上市放行标准要求后,才可以上市销售。[15]
结语
药品上市许可转让到实现商业化生产销售的过程中,存在诸多细节需要交易各方逐一敲定,对整体安排的构思只是交易的第一步,但跬步江山即寥廓。具体的合作模式如何体现在条款约定中、如何划分各方的权利义务关系、交易各方如何确保交易安排满足证券监管要求详见后续系列文章。
注释

